

Mišična distrofija
Mišična distrofija je grupa familiarnih bolezni, ki povzročijo degeneracijo skeletnih in mišičnih vlaken na osnovi genske mutacije. Poznamo nekaj tipov mišične distrofije. Dušenova mišična distrofija, Bekerjeva mišična distrofija in Miotonična mišična distrofija. Bekerjevo in Dušenovo distrofijo povzroči mutacija na istem distrofinskem genu medtem, ko mutacije na drugih genih povzroči druge vrste mišičnih distrofij. Pri teh drugi distrofijah proteini reagirajo s distrofin proteinom. Te druge mišične distrofije pa povzročajo podobne simptome, kot mišični distrofiji. (Dušenova in Bekerjeva) Gre za defekcijo mišic zaradi slabe prehranitve. To povzroči, da mišice postanejo šibke.
Pod mikroskopom je vidna sprememba same mišice. Ampak ne na živčnem sistemu. To razlikuje mišično distrofijo od drugih stanj, ki vsebujejo poškodbo živca. Na primer nevropatije. Pri slednji imamo poleg poškodovane mišice še poškodovan živec.
Dušenova mišična distrofija DMD
Podedovana bolezen, povzroči jo mutacija na distrofinskem genu. Na kromosomu X. Kadar je motnja tako velika, da ni proteina distrofina takrat nastopi DMD, ki je hujša oblika distrofije. Z simptomi, ki se pogosto pojavijo do 5 leta starosti.
Odsotnost distrofina omogoči, da skozi mišično celico vstopa prevelika količina kalcijevih ionov. Skozi mitohondrije vdrejo molekule vode, kar povzroči njihov propad. Propad mitohondrijev aktivira signale, ki povzročijo reakcijo katera povzroči poškodbo mišičnih celic in s tem propad mišičnih vlaken, le te pa začne nadomeščati maščobno in mehko tkivo. Tako se v telesu začne manjšati mišična masa. Povzroča degeneracijo mišičnega tkiva in ščasoma smrt.
Ob laboratorijskem testiranju se lahko mutacija določi že pri rojstvu. Zaradi izgubljanja mišične mase se prvi znaki pojavijo pri mišični slabosti v predelu medenice in v proksimalnih delih spodnjih okončin. Ta slabost se s časoma prenese na zgornje dele udov. V zgodnjem stadiju bolezni se začnejo pojavljati težave s hojo, psevdohipertrofija (povečanje mečnih mišic, deltoideusa in jezika), težave z vzdržljivostjo. V kasnejšem stadiju bolezni se mišično tkivo začne nadomeščati z maščobnim in mehkim tkivom. Tako, v 10-tem letu starosti bolnik že potrebuje bergle nato tudi te niso več dovolj, kar povzroči da bolnik pristane vezan na invalidski voziček. Degeneracija mišičnih vlaken in njihovo zmanjšanje mase, prepelje do nezmožnosti gibanja udov in do paralize.
Otroci z Dušenovo distrofijo shodijo kasneje v otroštvu. Imajo gegajočo hojo. Njihove mišice so povečane zaradi maščob, fibroze in ne zaradi mišic. Drugi klasični znak dušenove distrofije je Gowerjev znak. Če se otrok uleže na trebuh in poizkuša počasi vstati z uporabo svojih rok bo le to storil stežka, zaradi slabih zgornjih mišic ter na bokih in nogah. Kasnejši simptomi vključujejo potrebo po invalidskem vozičku zaradi resne oslabitve, pridelavo dihalnih težav zaradi šibkega diaframa, skolioza, (vstran ukrivljena hrbtenica) razširjena kardiomiopatija in aritmija. Distrofinski protein se nahaja tudi v srčni mišici. Na žalost te komplikacije vodijo v krajšo življensko dobo.
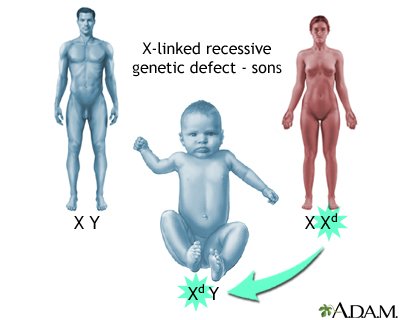
Ugotovljeno je, da se bolezen prenaša spolno na kromosomu X. Prenašalke oziroma nosilke so ženske, obolevajo pa moški. Bolezen se, kadar gre za mutiranje dveh kromosomov X, lahko pojavi tudi pri ženskah.
Stroka še ni razvila zdravila za DMD. Raziskave potekajo v smeri metode zdravljenja, ki onemogoča oziroma vsaj deloma omogoča nastanek proteina distrofina katerega pomanjkanje v celoti je ravno nastanek DMD.
Bekerjeva mišična distrofija BMD
Podedovana bolezen, povzroči jo mutacija na distrofinskem genu. Kadar mutacija ni tako velika, da proteina distrofina ni temveč, da se le ta preoblikuje govorimo o Bekerjevi mišični distrofiji. Gre za milejšo obliko dušenove distrofije, ki se pojavi kasneje. Običajno med 10 in 20 letom starosti.
Distrofinski gen je velik gen na X-Kromosomu. Ima 79 exonov v primerjavi z ostalimi geni, ki imajo 10 exonov. Več eksonov pomeni, da je večja verjetnost za mutacije med mejoso. Postopkom, kjer nastaneta sperma ali jajčece. Večina tih mutacij so izbris ali duplikacija enega ali več exonov. Manj je točkovnih mutacij.
Moški imamo en X in en Y kromosom. Ženske imajo dva X kromosoma. To pomeni, da je mišična distrofija pogostejša pri fantkih. Saj imajo le eno kopijo distrofinskega gena in, če je ta kopija defektivna je ta edina na razpolago mišičnim celicam. Medtem, ko ženske z defektivnim distrofinskim genom imajo lahko še drugega zdravega na razpolago. Ker sta obe distrofiji in Bekerjeva in Dušenova povezani z X kromosomom ju imenujemo X-Linked recessive.
Pri ženskah tako pride do izraza le en X kromosom, drug je pa deaktiviran. Kadar je ta deaktivacija naključna lahko pričakujemo, da ima polovica ženskih celic funkcionalen distrofinski gen in druga polovica defektnega. Ti ljudje so po navadi asimptomatski. Če torej več celic konča z defektnim distrofinskim genom in manj z funkcionalnim lahko postanejo manifestirani nosilci. Pomeni, da kažejo določene simptome.
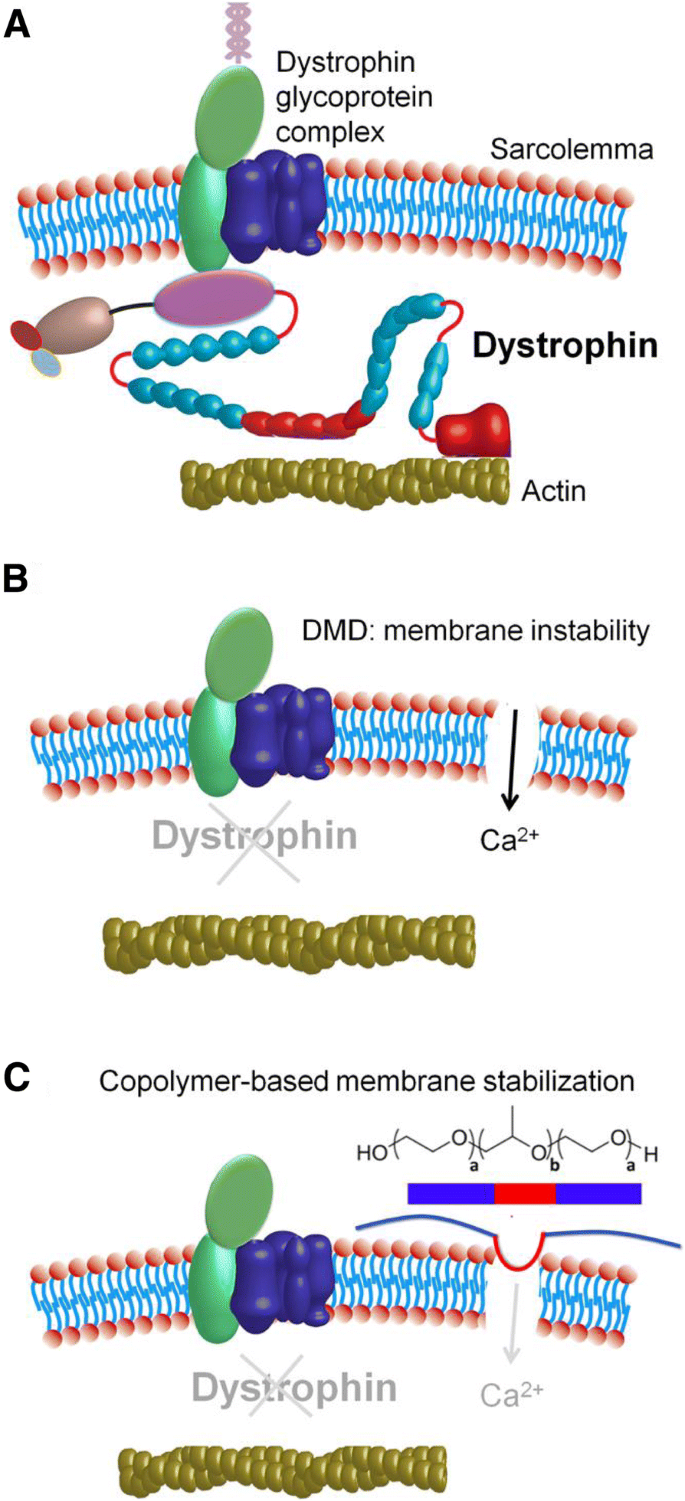
Distrofinski protein povezuje medcelični aktin z distrofinskim proteinskim kompleksom. Le ta je skupek citoplazmičnih in membranskih celičnih proteinov, ki je povezan z zunanjo celično matrico okoli celične mišice. Ta celotna povezava med aktinom in celično matrico stabilizira mišično celično membrano. Brez podpornega distrofina proteina na mestu mišična celična membrana postane šibka in nestabilna. S časoma celični proteini, kot so kreatin začno bežati iz poškodovane celice in kalcij začne vstopati v to celico. Celična membrana ne opravlja več svoje funkcije. To pa vodi do odmrtja celice.
Mišice se tudi hkrati regenerirajo, kar pa privede do različne velikosti in oblike mišice, kjer imamo na eni strani odmrle dele mišice in na drugi regenerirane. Ampak na dolgi rok imamo mišično atrofijo, ko v mišici nastajajo maščobe in fibrotično tkivo, kar oslabi mišico. Ta proces se še posebej opazi na nogah.
Na žalost stroka še ni razvila tretmaja za zdravljenje Dušenove ali Bekerjeve mišične distrofije. Glucocorticoidi lahko upočasnijo degeneracijo ampak lahko povzročijo tudi stranske učinke, kot npr povečana telesna teža. Drugi tretmaji, kot na primer fizična terapija in pogojevanje lahko izboljša kvaliteto življenja ampak ne more obrniti procesa.
Vedenje, razumevanje genetskega dedovanja, pogovor z starši osebe z distrofijo, razumevanje rizika pri naslednjem otroku z takim stanjem je pomembno. V 2/3 primerov je prenašalec otrokova mama in v drugi 1/3 je bolezen spontana. Kar pomeni, da jo povzroči nova mutacija. Če je nosilec mama in gledamo bodoče sinove polovica njih bo imela mutacijo. Kadar pa gledamo bodoče hčerke polovica od njih bo nadaljnjih prenašalcev mutacije.
Protein distrofin je zelo pomemben pri stabilizaciji celične membrane. Mutacije v genu lahko vodijo k izgubi tega proteina kar vodi v dušenevo mišično distrofijo. Pri mutacijah, kjer pride do popačitve distrofina pa vodi do Bekerjeve mišične distrofije.
Miotonična mišična distrofija
Pri tej vrsti distrofije govorimo o slabo prehranjenih mišicah, kar vodi v njihovo šibkost. Hkrati so te mišice skrčene, napete in jih ni mogoče sprostiti. Ravno tako, kot pri zgoraj opisanih distrofijah gre tudi pri miotonični mišični distrofiji za družino distrofij, popačenj oz. motenj. Nastane zaradi napake pri mutaciji v genih. Pomeni, da bolezen lahko povzroči že ena sama kopija gena.
Pojavlja se v vseh generacijah. Tako moški, kot ženska jo lahko preneseta naprej na otroka.
Poznamo dva tipa miotonične distrofije.
TIP 1 (DM1) Steinerjeva bolezen
TIP 2 (DM2)
TIP 1 Steinerjeva bolezen.
Prizadetost se pojavi na daljšem DNA genu. Kromosom 19. Le ta gen ima zgradbo, kateri se reče trinukleotidna ponovitev. To pomeni, da se 3-je DNA nukleotidi pojavijo po večkrat zaporedoma. Nukleotidi, ki sestavljajo ta gen so: Citozin, Timin in Guanin. Ti trije se pojavljajo v večkratnem zaporedju. Prizadetost se zgodi na koncu DNA gena, nastanek mRNA ampak ne proteina. Le ta mRNA na kromosomu 19 se nato transformira v miotonični protein kinase. Le ta pa pomaga pri medsebojni komunikaciji mišičnih celic. Ravno tako pomaga pri medsebojni komunikaciji med možganskimi celicami in medsebojni komunikaciji med srčnimi celicami.

V mišiči miotonični protein deluje tako, da ugasne miozin fosfatas. Slednji je pomemben pri krčenju, napenjanju in sprostivi mišice.
Poznamo 2 obliki Steinerjeve distrofije. Kongenitalna oblika, kjer se simptomi pojavijo že ob rojstvu, ko so dojenčki prešibki za dihanje ali sesanje. Ti dojenčki ne živijo dolgo. Nato imamo še odraslo obliko. Pri njej se pa šibkost mišic pojavi kasneje v življenju. Primarno prizadane obrazne mišice. Luknje na licih in padec vek. Mišice na spodnjem delu roke in mišice na spodnjem delu noge.
TIP 2
Prizadeti gen se pojavi na daljšem delu kromosoma 3. Ta gen ima zgradbo, ki se ji reče tetranukleotidna ponovitev. To pomeni, da se nukleotidi: Citozin, Citozin, Timin in Guanin pojavijo po večkrat zaporedoma. Ti nukleotidi se nahajajo v prvem intronu kromosoma 3. Intron je nekodirajoč odsek DNA znotraj gena. Ponovno govorimo o delu gena, ki je spremenjen v mRNA ampak ne v protein. Tu se ta del kromosoma 3 mRNA transformira v celično nukleidno kislino v vezalni protein. Ta pa kontrolira različne gene v mišicah in srcu.
Pri obeh tipih miotonične distrofije imamo ponavljajočo razširitev. Kjer imamo povečano število ponavljajočih zaporedij nukleotidov v prizadetih genih. Ta ponavljajočo razširitev povzroči zdrs pri združevanju nukleotidov. Rekombinantna DNA izgubi konsistenco se vrne in ponovno skopira kar je že prekopirala. Na mesto, da so nukleotidi združeni v daljšem zaporedju, se začne kopirati krajše zaporedje njihove združitve.
Ko se razvije plod pa dojenček in oseba v odraslo osebo. Preden se razvijeta sperma ali jajčece imamo že nešteto celic, ki vsebujejo takšno zaporedje zapisa. Več, kot je bilo dodanih kopiranih zapisov bolj nestabilno imamo strukturo.
Ta ekspanzija pomeni, da ima otrok enega od staršev z miotonično distrofijo (kopiranih nukleotidov) v sebi še več kopiranih nukleotidov oziroma takšnih genov od njegovega starša. Večje, kot je število kopiranih ponovitev prej v mladosti bo oseba začela kazati simptome miotonične distrofije in bolj resni bodo ti simptomi.
Zaradi kopije ponovitev v združevanju nukleotidov pride do napake združevanja nukleotida citozina in guanina. Tu se DNA vijačnica formira v kepice. To pa povzroči, da drugi geni niso pravilno formirani.
Miotonična tistrofija tip 2 se za razliko od tipa 1, pojavi le v odraslosti. Šibkost mišic je dosti bol blaga. Prizadane pa večinoma mišice v bokih in stegnih. Kar otežuje hojo po stopnicah in vstajanje iz sedečega položaja. Prizadane tudi ramena in komolce, kar povzroči težave pri držanju in dvigovanju predmetov.

Dobimo težave pri razvoju drugih genov za: Vid, težave pri genu za insulin in težave troponin gena srčne mišice.
Simptomi miotonične distrofije varirajo glede na tip miotonične distrofije. Obe mišični distrofiji povzročata miotonijo.
Miotonija je težava pri sproščanju mišic.
Osebe imajo lahko neodzivnost na insulin. To pomeni, da se celice ne odzivajo na insulin, ko transportira glukozo iz krvnega obtoka v tkivo. Kar pa vodi v povišano glukozo v krvi.
Osebe imajo lahko defekt pri krčenju srčne mišice. Ali pa abnormalije pri kontroli električnih signalov, ki kontrolirajo bitje srca.
Miotonična distrofija se diagnosticira z genskim testiranjem, kjer se preverja število ponovitev glukoidov v genu. Lahko se izvede tudi elektromiografija, kjer se preveri električna aktivnost mišic. Izvaja se tudi mišična biopsija, kjer se lahko razloči ali gre za mišični problem ali pa za živčni problem. Vse te diagnoze so zelo pomembne za informiranje posameznika glede možnosti prenosa bolezni na svoje potomce.
Miotonična distrofija se zdravi tako, da se zdravijo njeni simptomi. To lahko vključuje tudi uporabo bergl, podpornih elementov ali pa invalidskega vozička. Lahko tudi z operacijo in uporabo srčnega vspodbujevalnika, če je potreben.